Protein Folding & Discovery
Youtube search... ...Google search
- Life~Meaning ... Consciousness ... Creating Consciousness ... Quantum Biology ... Orch-OR ... TAME ... Proteins
- Symbiotic Intelligence ... Bio-inspired Computing ... Neuroscience ... Connecting Brains ... Nanobots ... Molecular ... Neuromorphic ... Evolutionary/Genetic
- Case Studies
- COVID-19
- Service Capabilities
- Architectures for AI ... Enterprise Architecture (EA) ... Enterprise Portfolio Management (EPM) ... Architecture and Interior Design
- (Deep) Residual Network (DRN) - ResNet
- RoseTTAFold: Accurate protein structure prediction accessible to all | Institute for Protein Design - University of Washington
- AI Reveals Previously Unknown Biology – We Might Not Know Half of What’s in Our Cells | University of California-San Diego
- Artificial intelligence powers protein-folding predictions | Michael Eisenstein - Nature
- Artificial intelligence has worked out the structures of 200 million proteins (that’s practically all of them) | Stephanie Pappas - Livescience
Proteins are made up of hundreds or thousands of amino acids, and these amino acid sequences specify the protein's structure and function. But understanding just how to build these sequences to create novel proteins has been challenging. Past work has resulted in methods that can specify structure, but function has been more elusive. Machine learning reveals recipe for building artificial proteins | Emily Ayshford - University of Chicago - PhysOrg
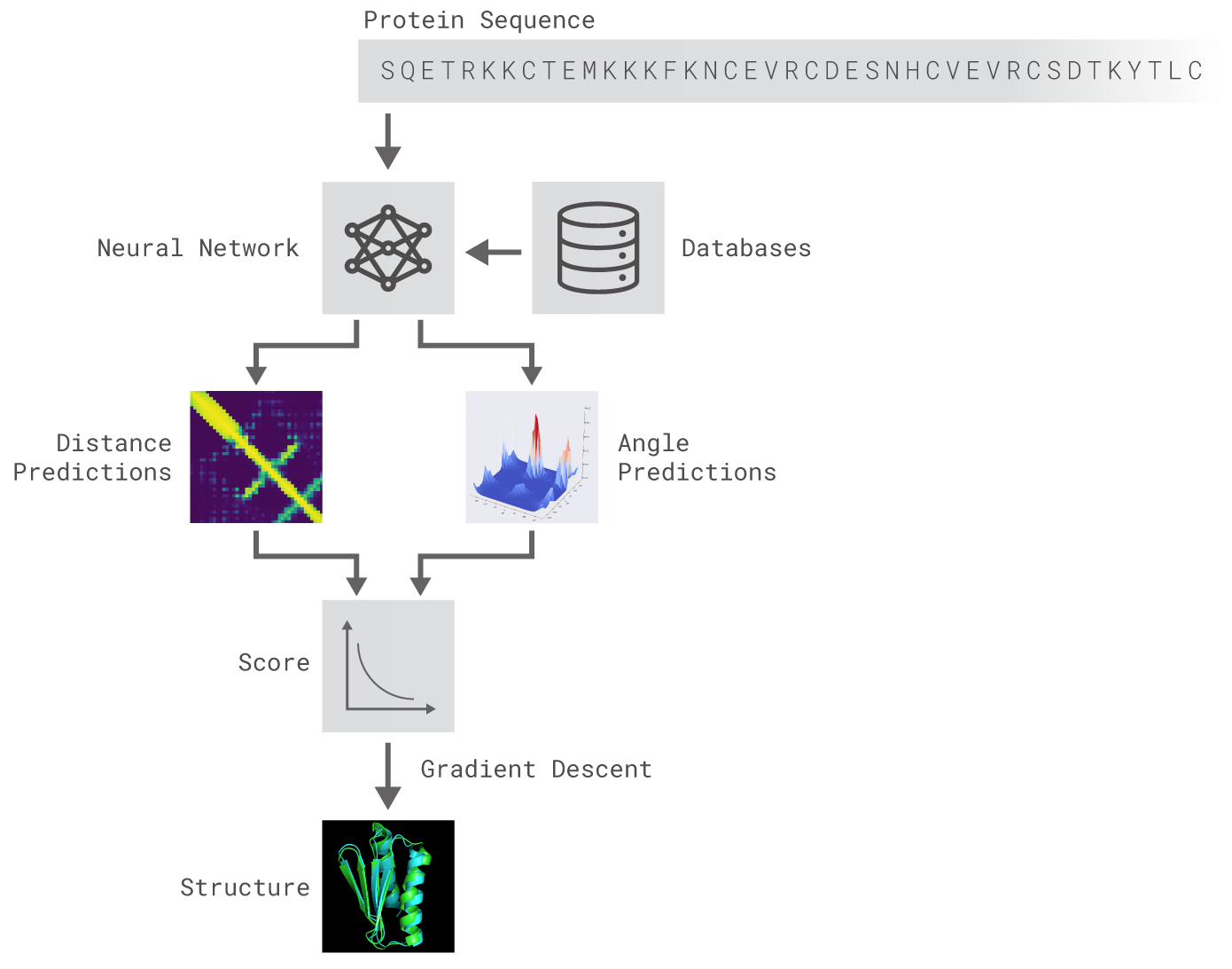
What is the protein folding problem? Proteins are large, complex molecules essential to all of life. Nearly every function that our body performs—contracting muscles, sensing light, or turning food into energy—relies on proteins, and how they move and change. What any given protein can do depends on its unique 3D structure. For example, antibody proteins utilised by our immune systems are ‘Y-shaped’, and form unique hooks. By latching on to viruses and bacteria, these antibody proteins are able to detect and tag disease-causing microorganisms for elimination. Collagen proteins are shaped like cords, which transmit tension between cartilage, ligaments, bones, and skin. Other types of proteins include Cas9, which, using CRISPR sequences as a guide, act like scissors to cut and paste sections of DNA; antifreeze proteins, whose 3D structure allows them to bind to ice crystals and prevent organisms from freezing; and ribosomes, which act like a programmed assembly line, helping to build proteins themselves. The recipes for those proteins—called genes—are encoded in our DNA. An error in the genetic recipe may result in a malformed protein, which could result in disease or death for an organism. Many diseases, therefore, are fundamentally linked to proteins. But just because you know the genetic recipe for a protein doesn’t mean you automatically know its shape. Proteins are comprised of chains of amino acids (also referred to as amino acid residues). But DNA only contains information about the sequence of amino acids–not how they fold into shape. The bigger the protein, the more difficult it is to model, because there are more interactions between amino acids to take into account. AlphaFold: Using AI for scientific discovery | A. Senior, J. Jumper, D. Hassabis, and P. Kohli - DeepMind
Contents
Google DeepMind AlphaFold
Youtube search... ...Google search
- Google DeepMind AlphaFold
- Google DeepMind AlphaGo Zero ... * Google DeepMind AlphaStar
- 13th Community Wide Experiment on the Critical Assessment of Techniques for Protein Structure Prediction
- Protein Data Bank | Wikipedia
- AlphaFold: a solution to a 50-year-old grand challenge in biology | DeepMind
- AI has cracked a problem that stumped biologists for 50 years. It’s a huge deal. | Sigal Samuel - Vox
- DeepMind and European Molecular Biology Laboratory (EMBL) release the most complete database of predicted 3D structures of human proteins | SciTechDaily Partners use AlphaFold, the AI system recognized last year as a solution to the protein structure prediction problem, to release more than 350,000 protein structure predictions including the entire human proteome to the scientific community.
- Highly accurate protein structure prediction with AlphaFold | J. Jumper, R. Evans, D. Hassabis - Nature
- FastFold: Reducing AlphaFold Training Time from 11 Days to 67 Hours | S. Cheng, R. Wu, Z. Yu, B. Li, X. Zhang, J. Peng, Y. You
- Google DeepMind Introduces AlphaFold 3: A Revolutionary AI Model that can Predict the Structure and Interactions of All Life’s Molecules with Unprecedented Accuracy | Asif Razzaq - MarketTechPost
DeepMind has brought together experts from the fields of structural biology, physics, and machine learning to apply cutting-edge techniques to predict the 3D structure of a protein based solely on its genetic sequence. AlphaFold: Using AI for scientific discovery | DeepMind
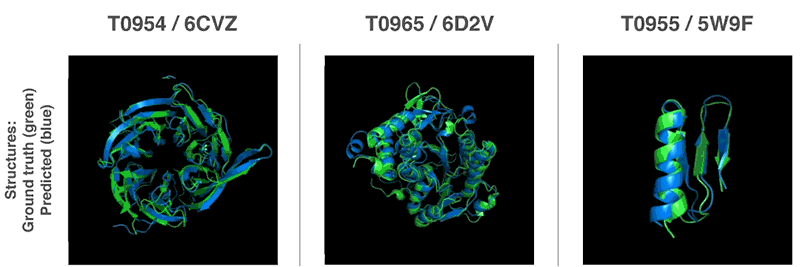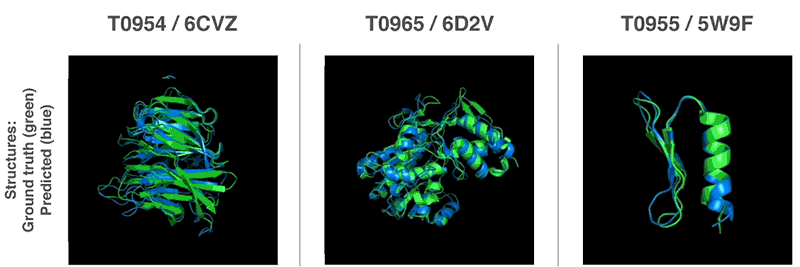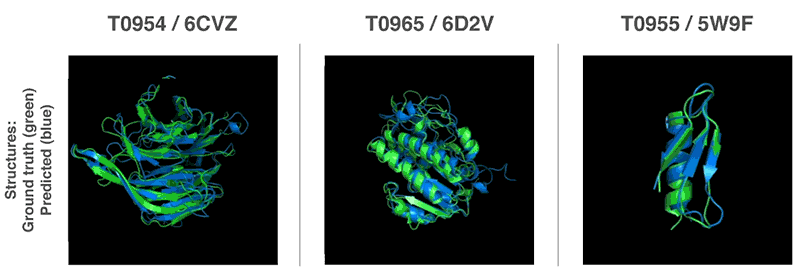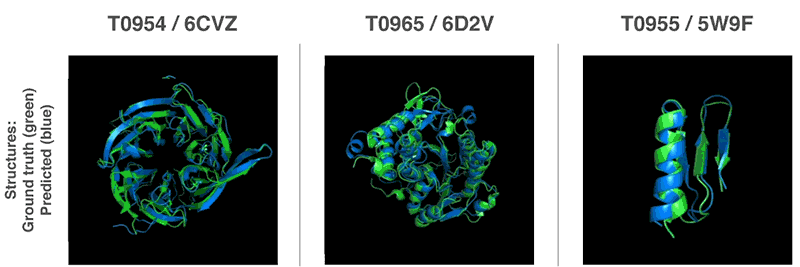
AlphaFold Demo
Google DeepMind and Isomorphic Labs have announced that scientists will have free access to most features of their newly launched research tool, the AlphaFold Server.
To use the AlphaFold server demo, you need to start by accessing the AlphaFold server website: https://AlphaFoldServer.com
Once there, you may need to create an account or log in if you haven't already.
Next, prepare your input data by creating a FASTA file that contains the amino acid sequence of the protein you wish to analyze. The FASTA format is a simple text-based format for representing nucleotide or peptide sequences, where each sequence is preceded by a description line starting with a ">" character. Make sure your FASTA file is correctly formatted and accurately represents the protein sequence.
Once your FASTA file is ready, go to the web interface of the AlphaFold server. Here, you will find an option to upload your FASTA file, usually through a clear "Upload" button. After uploading the file, you may need to provide additional details about the protein or adjust specific parameters for the prediction. This step ensures that the AI model has all the necessary information to process your job accurately.
After submitting the FASTA file and any necessary information, you can start the prediction job by clicking the button to begin the process, typically labeled "Submit," "Run," or something similar. The processing time can vary depending on the complexity of the protein and the current server load. You might see a progress bar or receive a notification once the job is complete.
When the prediction is finished, the server will provide an overview of the protein structure as a 3D rendering. This rendering is usually interactive, allowing you to manipulate and explore the 3D model directly within your web browser. Additionally, there should be options to download the predicted structure in various formats, such as PDB, for further analysis or use in other bioinformatics tools.
Finally, you can utilize the predicted structure for your research purposes. This might involve conducting further computational analysis, planning laboratory experiments, or integrating the structure into scholarly work. The streamlined process offered by the AlphaFold 3 server demo makes it significantly easier for researchers to obtain high-quality protein structure predictions and apply these insights to their scientific endeavors.
In summary, using the AlphaFold 3 server involves accessing the web interface, preparing and uploading a FASTA file, submitting the job, waiting for the prediction to complete, and then reviewing and utilizing the results. This user-friendly approach enables researchers to conduct sophisticated protein structure analysis with minimal effort, supporting a wide range of biological and biomedical research projects.
Baker Lab
- Gaming ... Game-Based Learning (GBL) ... Security ... Generative AI ... Games - Metaverse ... Quantum ... Game Theory ... Design
- Baker Lab
David Baker is a Nobel Prize-winning biochemist known for pioneering computational protein design, primarily through the Rosetta software suite he developed, which predicts protein structures and designs novel ones, enabling new medicines and enzymes by creating proteins from scratch to solve problems nature hasn't. His lab's innovations include:
- Rosetta Software: The foundational computational platform for protein structure prediction and design, initially developed in his lab, now a large collaborative community (Rosetta Commons).
- Protein Design: Creating new proteins with desired functions, moving beyond nature's limitations for medicine, catalysts, and sustainable solutions, earning him the 2024 Nobel Prize in Chemistry.
- RoseTTAFold & RFdiffusion: AI tools that use deep learning to rapidly and accurately predict protein structures, democratizing structure determination.
- Foldit: A game that leverages human puzzle-solving skills for protein folding challenges, engaging the public in scientific research.
Impacts:
- Revolutionized Biology: His work transformed protein design from fringe science to a powerful engineering discipline, allowing creation of molecules for therapies (like antivirals) and materials.
- Open Science: Fosters collaboration through the Rosetta Commons and open-source tools, accelerating discovery.
FASTA & FASTQ
FASTA and FASTQ are two widely used file formats in bioinformatics for storing nucleotide or peptide sequences. Here’s a detailed explanation of each:
FASTA is used to represent nucleotide sequences or protein sequences. The structure of a FASTA file includes a header line that begins with a '>' character followed by a description (e.g., sequence name or identifier), and sequence lines that contain the actual nucleotide or protein sequence in a single-letter code, which can span multiple lines. For example:
``` >sequence_1 ATGCGTAACGTAGCTAGCTAGCTAGCTA GCTAGCTAGCATCGATCGATCGATCGA ```
FASTQ is used to store both nucleotide sequences and their corresponding quality scores, which indicate the confidence of each base call, commonly produced by high-throughput sequencing technologies. The structure of a FASTQ file includes a header line that begins with an '@' character followed by a description (e.g., sequence name or identifier), a sequence line that contains the actual nucleotide sequence, a plus line with a '+' character (sometimes followed by the same identifier as in the header line, optionally), and a quality line with encoded quality scores corresponding to each nucleotide in the sequence line. The length of the quality line matches the length of the sequence line. For example:
``` @sequence_1 ATGCGTAACGTAGCTAGCTAGCTAGCTA + IIIIIIIIIIIIIIIIIIIIIIIIIIII ```
The key differences between these formats are that FASTA stores only the sequence information, while FASTQ stores both sequence information and quality scores. FASTA is typically used for sequence alignment, database searches, and various other bioinformatics analyses due to its simplicity and lower data intensity compared to FASTQ. On the other hand, FASTQ is essential in next-generation sequencing (NGS) workflows where quality scores are critical for downstream processing like read mapping, variant calling, and quality filtering, making it more complex due to the inclusion of quality scores.
Quality scores in FASTQ files are typically encoded using ASCII characters, with different versions of FASTQ (e.g., Sanger, Illumina 1.8+) using different offsets for encoding these scores. For instance, the Sanger format uses ASCII 33-73 to represent Phred quality scores ranging from 0 to 40. Understanding these formats is crucial for tasks such as sequence assembly, annotation, and various genomic analyses in bioinformatics.
CRISPR
Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) is a family of DNA sequences found in the genomes of prokaryotic organisms such as bacteria and archaea. It is the basis for the CRISPR-Cas9 technology used widely for gene editing. CRISPR acts as a bacterial immune system that uses RNA to guide a protein (most commonly Cas9) to detect and destroy DNA from invading viruses. Scientists adapted this mechanism to cut and edit DNA sequences in other organisms.
Significance
- Size: The most common effector protein, Cas9, is relatively large and bulky, which can sometimes make it difficult to package into standard viral vectors (like AAV) for delivery into cells.
- Precision: CRISPR-Cas9 requires a specific "flag" on the DNA (called a PAM sequence) to bind. While highly effective, this requirement restricts the number of targetable sites in the genome.
- Mechanism: It typically uses a single-guide RNA (sgRNA) to direct the Cas9 nuclease to a specific target site, where it induces a double-strand break in the DNA.
TIGR
Tandem Interspaced Guide RNA (TIGR) is a newly discovered RNA-guided system that targets DNA. It is currently being researched as a potential successor or alternative to CRISPR for gene editing. TIGR is a system found in bacteria and viruses that uses RNA to guide a protein (called Tas) to a specific DNA sequence to cut or modify it.
Significance
- Size: The proteins used (Tas) are significantly smaller (about 1/4 the size) than the Cas9 protein used in CRISPR, making them easier to deliver into cells for therapies.
- Precision: Unlike CRISPR, which requires a specific "flag" on the DNA (called a PAM sequence) to bind, TIGR systems do not require this, theoretically allowing them to target any sequence in the genome.
- Mechanism: It often uses a "dual-guide" approach (binding to both strands of the DNA helix), which may reduce off-target errors.
Nobel Prize: Protein Folding
Here is a list of Nobel Prizes directly related to the mechanisms, prediction, and determination of Proteins:
| When | Who | Prize Field | What |
|---|---|---|---|
| 1902 | Emil Fischer | Chemistry | For his work on sugar and purine syntheses. (Proposed the "peptide bond" theory, establishing that proteins are chains of amino acids linked together, and synthesized the first peptides.) |
| 1946 | James B. Sumner, John Northrop, and Wendell Stanley | Chemistry | For the discovery that enzymes can be crystallized and for the preparation of enzymes and virus proteins in a pure form. (Definitively proved that enzymes are proteins and that they have specific, defined chemical structures.) |
| 1948 | Arne Tiselius | Chemistry | For his research on electrophoresis and adsorption analysis. (Developed methods to separate complex mixtures of proteins, such as serum proteins in blood, laying the groundwork for protein analysis.) |
| 1954 | Linus Pauling | Chemistry | For his research into the nature of the chemical bond and its application to the elucidation of the structure of complex substances. ( Discovered the Alpha Helix and Beta Sheet, the two fundamental building blocks of protein secondary structure.) |
| 1958 | Frederick Sanger | Chemistry | For his work on the structure of proteins, especially that of insulin. ( [Image of insulin amino acid sequence] Determined the first complete amino acid sequence of a protein (insulin), proving proteins have a defined chemical composition.) |
| 1962 | Max Perutz and John Kendrew | Chemistry | For their studies of the structures of globular proteins. (Used X-ray crystallography to determine the first 3D atomic structures of proteins: hemoglobin and myoglobin.) |
| 1972 | Christian Anfinsen | Chemistry | For his work on ribonuclease, especially concerning the connection between the amino acid sequence and the biologically active conformation. (Established that the 3D structure of a protein is determined solely by its amino acid sequence.) |
| 1972 | Stanford Moore and William Stein | Chemistry | For their contribution to the understanding of the connection between chemical structure and catalytic activity of the active centre of the ribonuclease molecule. (Worked alongside Anfinsen to define the chemical structure of a protein's active site.) |
| 1984 | Bruce Merrifield | Chemistry | For his development of methodology for chemical synthesis on a solid matrix. (Invented solid-phase peptide synthesis, allowing researchers to build custom protein chains in the lab.) |
| 1988 | Johann Deisenhofer, Robert Huber, and Hartmut Michel | Chemistry | For the determination of the three-dimensional structure of a photosynthetic reaction centre. (First 3D structure of a membrane protein, which are notoriously difficult to crystallize compared to water-soluble proteins.) |
| 1992 | Edmond H. Fischer and Edwin G. Krebs | Physiology or Medicine | For their discoveries concerning reversible protein phosphorylation as a biological regulatory mechanism. (Discovered how proteins act as "switches," turning on and off via the addition of phosphate groups.) |
| 1994 | Alfred G. Gilman and Martin Rodbell | Physiology or Medicine | For their discovery of G-proteins and the role of these proteins in signal transduction in cells. (Identified the mechanism by which proteins transmit signals from the outside of a cell to the inside.) |
| 1997 | Stanley B. Prusiner | Physiology or Medicine | For his discovery of Prions - a new biological principle of infection. (Discovered that misfolded proteins alone can be infectious agents.) |
| 1997 | Paul D. Boyer, John E. Walker, and Jens C. Skou | Chemistry | For the elucidation of the enzymatic mechanism underlying the synthesis of ATP and the discovery of an ion-transporting enzyme. (Revealed the mechanical "motor" action of the massive protein complex ATP Synthase.) |
| 1999 | Günter Blobel | Physiology or Medicine | For the discovery that proteins have intrinsic signals that govern their transport and localization in the cell. (Discovered the "zip codes" embedded in amino acid sequences that direct proteins to their correct destination.) |
| 2002 | Kurt Wüthrich | Chemistry | For his development of nuclear magnetic resonance spectroscopy for determining the three-dimensional structure of biological macromolecules in solution. (Allowed scientists to see protein structures in their natural liquid state rather than just as crystals.) |
| 2003 | Roderick MacKinnon | Chemistry | For structural and mechanistic studies of ion channels. (Revealed how channel proteins permit specific ions (like Potassium) to pass through cell membranes while blocking others.) |
| 2004 | Aaron Ciechanover, Avram Hershko, and Irwin Rose | Chemistry | For the discovery of ubiquitin-mediated protein degradation. ( [Image of ubiquitin proteasome system] Discovered the "kiss of death" system where proteins are tagged with Ubiquitin to be destroyed and recycled.) |
| 2008 | Osamu Shimomura, Martin Chalfie, and Roger Tsien | Chemistry | For the discovery and development of the green fluorescent protein, GFP. ( Turned a jellyfish protein into a glowing tag that allows scientists to watch proteins move inside living cells.) |
| 2009 | Venkatraman Ramakrishnan, Thomas A. Steitz, and Ada E. Yonath | Chemistry | For studies of the structure and function of the ribosome. (Mapped the atomic structure of the Ribosome, the massive molecular machine that synthesizes proteins.) |
| 2012 | Robert Lefkowitz and Brian Kobilka | Chemistry | For studies of G-protein-coupled receptors. (Mapped the structure of GPCRs, the largest family of protein receptors and the target of nearly 50% of all modern drugs.) |
| 2013 | Martin Karplus, Michael Levitt, and Arieh Warshel | Chemistry | For the development of multiscale models for complex chemical systems. (Pioneered the computer simulations used to model protein dynamics.) |
| 2017 | Jacques Dubochet, Joachim Frank and Richard Henderson | Chemistry | For developing cryo-electron microscopy for the high-resolution structure determination of biomolecules in solution. (Revolutionized structural biology, allowing for high-resolution 3D models of proteins without crystallization.) |
| 2018 | Frances Arnold, George Smith, and Gregory Winter | Chemistry | For the directed evolution of enzymes (Arnold) and for the phage display of peptides and antibodies (Smith/Winter). (Used principles of evolution to breed new proteins with desired properties in the lab.) |
| 2021 | David Julius and Ardem Patapoutian | Physiology or Medicine | For their discoveries of receptors for temperature and touch. (Identified the specific Piezo and TRP proteins that mechanically sense heat and pressure.) |
| 2024 | David Baker, Demis Hassabis, and John Jumper | Chemistry | For computational protein design and protein structure prediction. (Solved the folding prediction problem with AI and enabled the creation of entirely new proteins from scratch.) |